Богатые азотом вещества, такие как отходы скотобоен и кожевенного производства, шерсть и т.д., могут служить не только сырьем для селитры, но и для получения цианистых соединений.
Самым простым способом переработки азотсодержащего сырья в цианистые соединения является получение желтой кровяной соли K4[Fe(CN)6]. Для этого высушенные отходы (обрезки кожи и меха, кровь, рога и копыта и т.д.) сплавляются с поташем и обрезками железа при высокой температуре, 900-1000 градусов. Сплавление можно производить на большой железной или чугунной сковороде, а лучше в толстостенной чугунной реторте, добавляя органический материал порциями к расплаву поташа с железом. Получающийся плав называется «синькали».
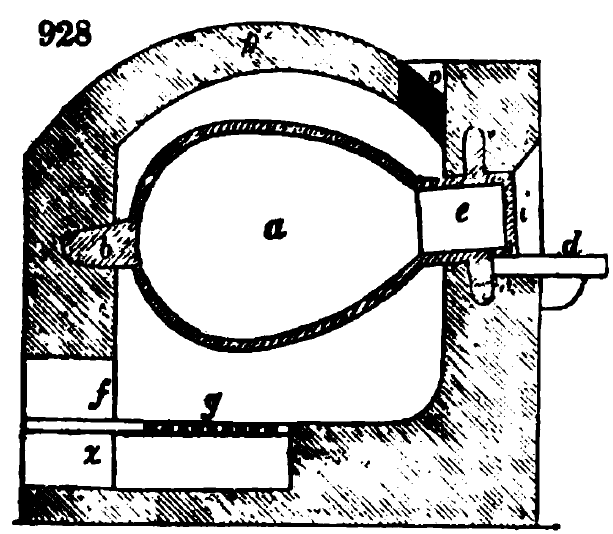
При сплавлении сначала происходит образование цианида калия, который, при дальнейшем действии на плав горячей воды реагирует с железом с образованием желтой кровяной соли, на что требуется около суток:
6KCN + Fe + 0.5O2 + H2O = K4[Fe(CN)6] + 2KOH
После фильтрации, упаривания растворов и кристаллизации выпадает желтая кровяная соль.
Поташа на каждую плавку берется около 100 кг, но в основном в виде упаренных маточных растворов от предыдущей плавки, свежего поташа из этого количества добавляется лишь 15-20 кг. Количество органического материала зависит от его качества, т.е. содержания азота, и обычно составляет 100-140 кг, а железных обрезков 5-10 кг на указанное количество поташа. В результате получается обычно 11-18 кг чистой желтой кровяной соли.
При сплавлении в цианид превращается 15-20% содержащегося азота, остальное улетает в виде аммиака, поэтому лучше сначала исходное сырье подвергнуть сухой перегонке, получая при этом аммиак (из 100 кг высушенного материала около 10-15 кг аммиака в виде аммиачной воды или карбоната аммония), и сплавлять с поташом уже получившийся животный уголь. А остаток от выщелачивания, в котором остается весь содержавшийся фосфор, годится как удобрение.
Исторически первое применение желтой кровяной соли состояло в приготовлении берлинской лазури (с 1704 г.), весьма ценного пигмента, при взаимодействии с солями железа (III):
3K4[Fe(CN)6] + 2Fe2(SO4)3 = Fe4[Fe(CN)6]3 + 6K2SO4
Стадию получения и выделения желтой кровяной соли при этом можно пропустить, и прямо добавить к раствору синькали соли железа.
Смесь желтой кровяной соли и бертолетовой соли, получающаяся совместной кристаллизацией из горячего раствора, и известная как «белый порох Ожандра», подходит в качестве ударного состава для капсюлей.
Кроме того, желтую кровяную соль можно окислить хлором или свинцовым суриком до красной кровяной соли K3[Fe(CN)6], которая пригодится, например, для светокопирования.
При взаимодействии гексацианоферрата натрия (для его получения нужно взять соду, а не поташ) с нитритом натрия образуется нитропруссид натрия Na2[Fe(CN)5NO], который с 1928 г. и по настоящий момент применяется в медицине как средство для быстрого понижения артериального давления при гипертоническом кризе или для уменьшения кровепотерь во время операций.
Из желтой кровяной соли легко получить цианиды калия и натрия, которые будут очень полезны для электрохимического золочения и серебрения, добычи золота из руд, цианирования стали, получения оргстекла и т.д.. Для этого желтую кровяную соль нужно осторожно расплавить в закрытом тигле, и после охлаждения извлечь цианид водой:
К4[Fe(CN)6] = 4KCN + FeC2 + N2
Но лучше добавить к желтой кровяной соли разбавленную серную кислоту и выделяющийся циановодород (помня о его токсичности) поглотить раствором щелочи:
K4[Fe(CN)6] + 3H2SO4 = 2K2SO4 + FeSO4 + 6HCN
HCN + NaOH = NaCN + H2O
Есть и другие способы получения цианидов, например, из аммиака и муравьной кислоты, из карбоната натрия, угля и аммиака (Beilby process, использовался в конце XIX — начале XX вв.), или даже напрямую из воздуха (Bucher process). Последний способ заслуживает отдельного обсуждения.
Статья по истории берлинской лазури в Коммерсанте
https://www.google.com/amp/s/www.kommersant.ru/amp/4190096
//Фриш продавал берлинскую лазурь по 30 талеров за фунт, что по покупательной способности денег того времени соответствует примерно 1000 современных евро. Это было не дешево, но и не дорого, потому у берлинской лазури на тот момент был только один реальный конкурент — натуральный ультрамарин, он же ляпис-лазурь.//
«При сплавлении в цианид превращается 15-20% содержащегося азота, остальное улетает в виде аммиака, поэтому лучше сначала исходное сырье подвергнуть сухой перегонке, получая при этом аммиак (из 100 кг высушенного материала около 10-15 кг аммиака в виде аммиачной воды или карбоната аммония), и сплавлять с поташом уже получившийся животный уголь. »
Вот тут непонятно. Как я понимаю, в исходном процессе азот белков частично вяжется с углеродом в присутствии щёлочи в цианид. Плюс, железо выводит цианид из равновесия.
А что получится в животном угле? В какой форме там азот останется?
Скорей всего, получаются какие-нибудь полициклические азотсодержащие гетероциклы, для этого достаточно 200-400 градусов, или циклические дипептиды, которые очень термически стойки.
Сам процесс образования цианида идет при более высокой температуре, около 1000. Поэтому имеет смысл или разделять стадии, получая дополнительно аммиак, или, как это иногда делалось, загружать смесь в вертикальную трубчатую реторту и начинать нагрев сверху — тогда аммиак и летучие азотсодержащие вещества проходят через уже накаленный слой угля с поташом и почти полностью связываются в цианид.
Весьма хитрым сырьём получается такой азотистый «животный уголь», и к температуре чувствительным — не пережарить, не недожарить… и к исходному сырью, возможно.
А из технологии с трубой следует возможность получения из мочи (в т.ч. загаженной земли или подстилки скота — промываем и скармливаем раствор печке понемногу). Или из мокрых отбросов, теряющих азот при сушке. Или из водяного отгона при пиролизе растений.
Это всё, подразумевая, что концентрированный сухой белок может быть в дефиците.
//Весьма хитрым сырьём получается такой азотистый «животный уголь», и к температуре чувствительным — не пережарить, не недожарить…//
Дак как раз нет, все, что легко выделяется, отгоняется до 300-400, а дальше почти ничего не отгоняется. В итоге весь азот или в угле, или в отгоне (аммиачная вода и масло).
Получение цианида из аммиака — это Beilby process, в конце статьи про него упоминается.
Нужно смотреть ценность готового продукта. Не думаю, что обрезки кожи не достать задаром или очень дешево. А вот горох или мясо никто переводить не даст))
//производство синъ-кали и может показаться крайне простымъ, тем не менее
получение его в химической технике сопряжено с очень большими затруднениями.
Вследствие тех многочисленных манипуляций,
с которыми сопряжено производство синь-кали. a
также и вследствие того, что подобными же манипуляциями пользуются и въ
других производствахъ, мы здесь подробнее с ними познакомимся и таким
образом при дальнейшем изложении будем иметь возможность ссылаться на здесь описамные методы работы.
Как мы только что видели, для приготовления желтой кровяной соли
можно употреблять всевозможные животные остатки, и таким образом это
производство для сельскаго хозяйства имеет особенное значение, утилизируя животные отбросы,
которые прежде являлись совершенно неценными.
В настоящее время известно, что все азот содержащие отбросы могут
быть употребляемы, как землеудобрительныя средства. Прежде этими
остатками пользовались исключительно для одной цели, а именно для
приготовления углеаммиачной соли, которая есть не что иное, как углекислый
аммоний. Перегонка животных отбросов сопровождается выделением изъ
них азота в виде аммиака, который и соединяется с выделяющейся
одновременно углекислотой в углекислый аммоний. Кроме того при перегонке
этих отбросов образуется неприятнаго запаха масло, известное под назваыием масла Диппеля.
Остатокъ, остающийся при этом в ретортахъ, представляет собою обугленную массу,
содержащую еще довольно много азота, и дает при сплавлении
с поташем кровещелочную соль. Химических процессовъ, происходящих при этом процессе,
мы будем касаться по мере описания производства. Таким образом заводы, приготовляющие углеаммиачную соль,
занялись производством синь-кали уже ради того, чтобы как-нибудь
съ пользою освободиться от этихъ уголъных остатковъ. В кристаллическомъ
виде эта соль появилась в продаже лишь в 20 годах прошлаго столетия,
до этого времени выходящий изъ печи сплав непосредственно перерабатывали
на берлинскую лазурь, способъ, соиряженный съ болъшим расходом поташа и не
дающий столь красивой краски, как она получается при употреблении очищенной желтой соли.
В средине прошлаго столетия добыванию аммиака или углеаммиачной соли
из животныхъ остатков был положен конецъ, когда в промывных водахъ
газовых заводов нашли более дешевый и неисчерпаемый источник для
получения аммиака. С этого же времени заводы, приготовляющие желтую
синильную соль, перестали обугливать животные остатки, такъ как
это стало невыгоднымъ, и перерабатывают их теперь в сыром виде.//
//Б. К Климов исследованием, проведенным (1924) в полузаводском масштабе
на опытном заводе Гос. Инст. прикл. химии, показал, что желтая соль может быть
получена с очень хорошим выходом при накаливании с железом и поташом богатых
азотом сапроколлов [ископаемых сапропелей (I т., стр. 476)1, находимых во многих болотах,
напр., в Толполовском, Детскосельского уезда Ленинградской губ. (Г.)//
//Цианистый калий, т. е. ея калийная соль, такъ-же ядовитъ, как она сама.
До средины XIX столетия цианистый
калий не находил большого применения даже в лабораторияхъ. Но съ
тех поръ, как выяснилось, что он в некоторых отношениях в тех-
нике незаменимъ, его стали употреблять в больших количествахъ. Почти
никогда не бывает несчастных случаевъ, которые могли бы быть вслед ствие
его сильной ядовитости, так как люди, имеющие с ним дело, благодаря
долголетней практике, умеют защищать себя от его вредныхъ
действий.//
Цианидный способ фиксации азота уже обсуждали (http://popadancev.net.s3-website-us-east-1.amazonaws.com/ammiak/#comment-149895), но тут попалась целая глава в книжке по этому вопросу
https://chem21.info/page/146015241074127171184073107069028244194049094143
Из книжки The worlds greatest fix. A history of nitrogen and agriculture
//Scheele probably observed that dinitrogen reacts with heated sodium carbonate and carbon as early as 1775, and in 1839 iron was observed to catalyse this reaction, which generates cyanides. One John Swindells took out British Patent 8036 in 1839 describing the manufacture of cyanides from a mixture of sodium or potassium sulfate, calcium oxide or carbonate, iron filings, and coal heated in a furnace in air. Associated products were carbonates and sulfides.
There are reports from 1813, 1819, and 1826 of lumps of fused salts that were to be found in the bottom of iron blast furnaces. These were assumed to be chlorides or carbonates, and it was only in 1835 that it was recognised that this kind of material also contained cyanide.
The phenomenon became more and more common as improved technology allowed the temperatures of operation of blast furnaces to increase. The combination of an extremely high temperature, a metal such as iron, carbon in the form of coal, and atmospheric nitrogen is just what one might now expect to produce cyanide. When new hot blast furnaces were introduced on the Clyde in Scotland in 1837, the workmen soon noticed “peculiar exudations of fused salt” forming upon the walls of the furnace. One particularly observant workman apparently noted that this material was alkaline and took it home to assist his wife in washing the family clothes, where it was apparently used as a substitute for soap. Chemical analysis later showed the material to be a mixture of potassium carbonate, K2CO3 (45.8%), and potassium cyanide, KCN (43.4%), together with other, possibly noxious materials.
Apparently, the furnace produced cyanide for at least three more years, but it is not recorded how long the workman’s wife survived to wash clothes, though Breneman says somewhat laconically that she used the material “for a time”. A German commentator of the period noted drily that the material was not suitable for domestic use.
Throughout the nineteenth century, there were reports of cyanide (and a related material, cyanate) being produced in blast furnaces. The production seems to have been commonly recognised, but it is now not clear whether this was regarded as a hazard, an advantage, or merely a nuisance. Breneman records having observed cyanide formation in blast furnaces on at least two occasions. Bunsen and Playfair reported in 1845 that a furnace at Alfreton, Derbyshire, in the north of England, yielded 224.7 lb (there were no SI units then, though in a footnote they express a preference for the “French” units of metres and grammes) of cyanide in 24 hours. It seems to have been produced in a very restricted part of the furnace, so temperature and gas composition were probably critical. The conclusion, after considerable discussion, was that hot carbon and atmospheric nitrogen could react to give cyanide and that, as potash was generally present, the final product was potassium cyanide. Water also played a critical role. If it was present, then cyanide was still a product, but the main compound then produced was cyanogen, which has the formula C2N2: 2C + N2 → C2N2/
//Either way, it was quite clear by 1850 that industrial fixing of nitrogen by this kind of route was feasible. However, the product, cyanide, was hardly going to be of direct use as a fertiliser, though one might have envisaged applications in dyeing (and, less benevolently, in dying!). An Anglo–French attempt to commercialise cyanide preparation seems to have started near Paris in 1843. In this endeavour, the production of potassium ferrocyanide (then termed yellow prussiate) reached about fifteen tons per year, but the initial compound formed was actually potassium cyanide. The process used dried charcoal that had previously been soaked in a solution of potassium carbonate, and this was heated white hot in clay retorts through which the exhaust gases from coal fires were passed. This was presumably to heat the dinitrogen of the air and remove dioxygen before introducing the gases into the furnace.
Since Britain was, at that time, truly the workshop of the world, the process was moved from Paris to Newcastle-upon-Tyne in 1844. There it was set up on a larger scale, producing yellow prussiate at the rate of more than a ton per day. The process was carried out in cylinders made of firebrick, ten feet long, two feet in diameter, and with walls nine inches thick. Air was admitted to them through layers of charcoal. The cyanide-bearing charcoal was dropped into tanks of water containing a suspension of spathic iron ore (siderite, essentially iron carbonate), and the solution of yellow prussiate thus produced was then evaporated to induce crystallisation.
This complicated process was only operated for two or three years, and it was abandoned in 1847. The cost of the salt was said to be less than 2 francs (French) per kilo, which translated then as about $0.20 (U.S.) or £0.05 per kilo, but this was not a commercial price. Part of the reason must have been that the retorts suffered from excessive corrosion and needed replacement after several months. The toll on the operators must also have been considerable, though this was not a pressing consideration of the time. Throughout the rest of the nineteenth century, attempts were made to develop an industrial process for the manufacture of cyanide that would also be a commercial success, but Breneman could write in 1889 that: “The history of later attempts to utilise the nitrogen of the air for the manufacture of cyanides, shows, up to the present time, no commercial success.”//
цианамид кальция CaNCN, который обычно производят по методу Франка-Каро из карбида кальция CaC2 и азота, является не только хорошим азотныи удобрением, но и сырьем для многих важных веществ — цианида натрия, гуанидина и его производных, дициандиамида, меламина и многих других.
Однако, при недоступности карбида кальция цианамид можно получать гораздо более простым путем, нагреванием смеси негашеной извести м мочевиной. При этом сначала, при температурах 150-250 градусов, происходит образование цианата кальция
2CO(NH2)2 + CaO = Ca(OCN)2 + H2O + 2NH3
который при более высокой температуре, 700-900 градусов, превращается в цианамид
Ca(OCN)2 = CaNCN + CO2
Чтобы получить из цианамида нитрат гуанидина, достаточно сплавить CaNCN с аммиачной селитрой при 95-100 градусах
CaNCN + 4NH4NO3 = CNH(NH2)2*HNO3 + Ca(NO3)2 + 2NH3
По сравнению с методом прямого получения нитрата гуанидина из мочевины и аммиачной селитры http://popadancev.net.s3-website-us-east-1.amazonaws.com/bezdymnyj-porox/#comment-153510 такой метод значительно безопасней и проще масштабируется.
Чтобы получить дициандиамид, цианамид кальция нужно купятить в воде, или же пропускать углекислый газ через суспензию CaNCN в воде
CaNCN + H2O + CO2 = H2NCN + CaCO3
2H2NCN = (H2N)2CNCN
Чтобы превратить CaNCN в цианид натрия, достаточно произвести сплавление с обычной солью и угольным порошком
CaNCN + C + 2NaCl = 2NaCN + CaCl2
Из мочевины с поташа или содой при нагреванит также получается цианат
3CO(NH2)2 + N2CO3 = 2NaOCN + 2CO2 + 4NH3
При более высоких температурах цианат натрия разлагается на цианид, карбонат, азот и CO. Однако в присутствии угля цианат полность восстанавливается в цианид, что может служить удобным методом его получения в небольших количествах
3NaOCN + 2C = 3NaCN + CO + CO2